| Crystal Structure Prediction and its Application in Earth and Materials Sciences |
| Crystal Structure Prediction and its Application in Earth and Materials Sciences |
To obtain further chemical insight into these exotic xenon oxides, we selected some of the stable structures containing Xe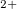 , Xe
, Xe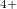 , and Xe
, and Xe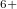 , namely, XeO-Pbcm at 100 GPa, XeO
, namely, XeO-Pbcm at 100 GPa, XeO -P2
-P2 /c at 150 GPa, and XeO
/c at 150 GPa, and XeO Pmmn at 200 GPa. Fig. 5.3 shows the density of states and its projection onto atomic orbitals. All of these xenon oxides are narrow-gap semiconductors. Using state-of-the-art GW calculations, we got the band gaps: 1.52 eV for XeO-Pbcm, 0.52 eV for XeO-P2/c, 0.15 eV for XeO-Pmmn; these values should be accurate to within 5-10%.
Pmmn at 200 GPa. Fig. 5.3 shows the density of states and its projection onto atomic orbitals. All of these xenon oxides are narrow-gap semiconductors. Using state-of-the-art GW calculations, we got the band gaps: 1.52 eV for XeO-Pbcm, 0.52 eV for XeO-P2/c, 0.15 eV for XeO-Pmmn; these values should be accurate to within 5-10%.
The highest valence band levels are dominated by p orbitals of O and Xe. Both the atom-projected densities of states and energy-decomposed electron densities suggest charge transfer from Xe to O atoms. Note that the contribution of Xe 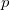 orbitals strongly decreases with increasing Xe oxidation because of two concurring effects, the change in stoichiometry and the parallel enhanced electron transfer from Xe to O. These two effects are disentangled in the DOS reported in Supplementary Material. The valence states dominated by p contributions contain in XeO-Pbcm about 40 electrons, with almost equal contributions from O and Xe. The contribution is almost all p, namely 4.51 p electron per Xe and 4.93 p electrons per O atom. In the case of XeO-P2/c there are about 112 electrons, 31.94 from Xe and 80.16 from O, which implies about 4 e per Xe and 5 e per O, which suggests that about one electron is transferred from a p-orbital of Xe into a p-orbital of O. Finally, in XeO Pmmn, there are about 45 electrons in the valence states dominated by p orbital contributions, 5.48 e from Xe and 30.10 e from O, leading to a further lowering to 2.8 electrons on Xe p orbitals and to again about 5 electrons on O p orbitals.
orbitals strongly decreases with increasing Xe oxidation because of two concurring effects, the change in stoichiometry and the parallel enhanced electron transfer from Xe to O. These two effects are disentangled in the DOS reported in Supplementary Material. The valence states dominated by p contributions contain in XeO-Pbcm about 40 electrons, with almost equal contributions from O and Xe. The contribution is almost all p, namely 4.51 p electron per Xe and 4.93 p electrons per O atom. In the case of XeO-P2/c there are about 112 electrons, 31.94 from Xe and 80.16 from O, which implies about 4 e per Xe and 5 e per O, which suggests that about one electron is transferred from a p-orbital of Xe into a p-orbital of O. Finally, in XeO Pmmn, there are about 45 electrons in the valence states dominated by p orbital contributions, 5.48 e from Xe and 30.10 e from O, leading to a further lowering to 2.8 electrons on Xe p orbitals and to again about 5 electrons on O p orbitals.
![\includegraphics[scale=1.0]{chapter5/pdf/fig3.png}](images/img-0267.png) -P2/c at 150 GPa, c) XeO-Pmmn at 200 GPa. The sum over the plotted projections gives the total DOS for each system. The DOS plotted have different units on the y-axis so as to give the total number of valence electrons for each system when integrated up to the Fermi level. This corresponds to 56
-P2/c at 150 GPa, c) XeO-Pmmn at 200 GPa. The sum over the plotted projections gives the total DOS for each system. The DOS plotted have different units on the y-axis so as to give the total number of valence electrons for each system when integrated up to the Fermi level. This corresponds to 56  (XeO, Z=4), 160 (XeO, Z=8) and 52 (XeO, Z=2).
(XeO, Z=4), 160 (XeO, Z=8) and 52 (XeO, Z=2).It is well known that DFT calculations systematically underestimate the band gap. To obtain accurate results, we performed GW calculations using the VASP code. Pressure dependences of the band gaps are shown in Fig. 5.4. All three compounds are semiconducting.
![\includegraphics[scale=0.7]{chapter5/pdf/bandgap.png}](images/img-0269.png)
The charge transfer issue was also investigated on the basis of the electron density using Bader’s analysis 166. For XeO-Pbcm the net charge on the Xe atoms is 1.01, while it increases to 1.995 on average in XeO-P2/c and to 2.754 in XeO-Pmmn. Note that the net charge on oxygen atoms stay almost the same in all the three compounds and very close to -1. This corresponds to ionicity of about 50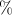 in all oxides. Note that the charge transfer described through the observable function, the electron density, and Bader’s analysis confirms what was found with using orbital-projected DOS, a totally independent approach.
in all oxides. Note that the charge transfer described through the observable function, the electron density, and Bader’s analysis confirms what was found with using orbital-projected DOS, a totally independent approach.
Bader analysis yields not only the net atomic charges, but also more subtle characteristics such as the three eigenvalues 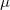
 (i=1,3) of the traceless quadrupole moment tensor Q(
(i=1,3) of the traceless quadrupole moment tensor Q( ), which is a good quantitative indicator of the departure of an atomic basin from sphericity. The eigenvectors associated with the eigenvalues give the principal directions for relative charge accumulation and depletion. For a spherical distribution the are all equal to zero. Deviation from zero indicates asphericity: negative eigenvalues arise from accumulation of charge in the direction associated with the corresponding eigenvector and at the expense of the directions associated with positive . Our calculation (Table 5.2) shows that Xe atoms in XeO-Pmmn are definitely more spherical, their being all close to zero and about one order of magnitude lower than for the Xe atoms in XeO and XeO. This agrees with the ELF picture while providing also a quantitative measure of sphericity. XeO-Pbcm (with one p orbital partially empty) has, in agreement with the orbital picture, one direction of relative charge depletion, associated to , and two directions of unequal relative charge accumulation, associated to
), which is a good quantitative indicator of the departure of an atomic basin from sphericity. The eigenvectors associated with the eigenvalues give the principal directions for relative charge accumulation and depletion. For a spherical distribution the are all equal to zero. Deviation from zero indicates asphericity: negative eigenvalues arise from accumulation of charge in the direction associated with the corresponding eigenvector and at the expense of the directions associated with positive . Our calculation (Table 5.2) shows that Xe atoms in XeO-Pmmn are definitely more spherical, their being all close to zero and about one order of magnitude lower than for the Xe atoms in XeO and XeO. This agrees with the ELF picture while providing also a quantitative measure of sphericity. XeO-Pbcm (with one p orbital partially empty) has, in agreement with the orbital picture, one direction of relative charge depletion, associated to , and two directions of unequal relative charge accumulation, associated to 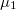 and
and 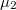 . Finally in XeO-P2/c, there is one direction of relative charge accumulation associated to and two of unequal relative charge depletion, associated to and
. Finally in XeO-P2/c, there is one direction of relative charge accumulation associated to and two of unequal relative charge depletion, associated to and 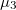 , in agreement with the picture of two p orbitals (partially) empty.
, in agreement with the picture of two p orbitals (partially) empty.
-P2/c at 150 GPa, and XeO-Pmmn at 200 GPa System |
|
|
|
XeO |
-3.49 |
-2.04 |
5.53 |
XeO |
-5.50 |
1.83 |
3.67 |
XeO |
-5.90 |
2.33 |
3.57 |
XeO |
-0.38 |
0.21 |
0.36 |
| Crystal Structure Prediction and its Application in Earth and Materials Sciences |