2 Methods
Structure searching for ground-state phases of Mg(BH )
) were performed with the ab initio evolutionary algorithm USPEX 4; 127. When nothing is known about the crystal structure, separate searches are usually done with increasing number of formula units in the unit cell – as long as computational resources allow and until the solution is found. We found structure solutions already in searches with 2 formula units (22 atoms/cell) and 4 formula units (44 atoms/cell), which yielded structures fully explaining experimental results. We did structure prediction at 0, 2, 5, 10, 15, and 20 GPa. All structures were relaxed by the VASP code. During structure searches, we used BH groups as whole units with fixed bond connectivity. All experimentally known structures contain these nearly rigid units at pressures below 22 GPa 128. This was done with the Z-matrix representation for the BH complex anions, keeping the bond connectivity within the BH tetrahedron, and fully relaxing the bond lengths and angles. This approach allowed us to focus searches on chemically interesting structures and reduced the search space dramatically 127. The presence of the same nearly rigid BH groups, connected to Mg atoms by weak bonds implies that zero-point energies of different structures will be nearly identical and thus can be safely neglected when comparing energetics of different structures; our calculations confirm this. We employed PAW-PBE functional for exchange and correlation. A cutoff energy of 600 eV, and a Monkhorst-Pack Brillouin zone sampling grid with the resolution of
were performed with the ab initio evolutionary algorithm USPEX 4; 127. When nothing is known about the crystal structure, separate searches are usually done with increasing number of formula units in the unit cell – as long as computational resources allow and until the solution is found. We found structure solutions already in searches with 2 formula units (22 atoms/cell) and 4 formula units (44 atoms/cell), which yielded structures fully explaining experimental results. We did structure prediction at 0, 2, 5, 10, 15, and 20 GPa. All structures were relaxed by the VASP code. During structure searches, we used BH groups as whole units with fixed bond connectivity. All experimentally known structures contain these nearly rigid units at pressures below 22 GPa 128. This was done with the Z-matrix representation for the BH complex anions, keeping the bond connectivity within the BH tetrahedron, and fully relaxing the bond lengths and angles. This approach allowed us to focus searches on chemically interesting structures and reduced the search space dramatically 127. The presence of the same nearly rigid BH groups, connected to Mg atoms by weak bonds implies that zero-point energies of different structures will be nearly identical and thus can be safely neglected when comparing energetics of different structures; our calculations confirm this. We employed PAW-PBE functional for exchange and correlation. A cutoff energy of 600 eV, and a Monkhorst-Pack Brillouin zone sampling grid with the resolution of 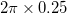
 was used. To examine the importance of long-range dispersion interactions on this compound’s thermodynamic stability, the semi-empirical dispersion-correction method was applied to the ground state structures 51. Phonon dispersion curves were calculated by using the super cell method as implemented in the PHONOPY code 67. The powder XRD patterns were simulated using the REFLEX software.
was used. To examine the importance of long-range dispersion interactions on this compound’s thermodynamic stability, the semi-empirical dispersion-correction method was applied to the ground state structures 51. Phonon dispersion curves were calculated by using the super cell method as implemented in the PHONOPY code 67. The powder XRD patterns were simulated using the REFLEX software.