| Crystal Structure Prediction and its Application in Earth and Materials Sciences |
| Crystal Structure Prediction and its Application in Earth and Materials Sciences |
 carbon allotropes from evolutionary metadynamics simulation :
Bibliography
carbon allotropes from evolutionary metadynamics simulation :
Bibliography
J Maddox. Crystals from first principles. Nature, 335:201, 1988.
A. Gavezzotti. Are crystal structures predictable? Acc. Chem. Res., 27:309–314, 1994.
A. R. Oganov(ed). Modern Methods of Crystal Structure Prediction. WILEY-VCH, Weinheim, 2010.
A. R. Oganov and C. G. Glass. Crystal structure prediction using evolutionary algorithms: principles and applications. J. Chem. Phys., 124:244704, 2006.
J. Pannetier, J. Bassas-Alsina, J. Rodriguez-Carvajal, and V. Caignaert. Prediction of crystal structures from crystal chemistry rules by simulated annealing. Nature, 346:343–345, 1990.
J. C. Schon and M. Jansen. First step towards planning of syntheses in solid-state chemistry: Determination of promising structure candidates by global optimization. Angew. Chem. Int. Ed. Engl., 35:1286–1304, 1996.
R. Martoňák, A. Laio, and M. Parrinello. Predicting crystal structures: The parrinello-rahman method revisited. Phys. Rev. Lett., 90:075503, 2003.
D. J. Wales and J. P. K. Doye. Global optimization by basin-hopping and the lowest energy structures of lennard-jones clusters containing up to 110 atoms. J. Phys. Chem. A, 101:5111–5116, 1997.
C. M. Freeman, J. M. Newsam, S. M. Levine, and C. R. A. Catlow. Inorganic crystal structure prediction using simplified potentials and experimental unit cells: application to the polymorphs of titanium dioxide. J. Mater. Chem., 3:531–535, 1993.
S Goedecker. Minima hopping: Searching for the global minimum of the potential energy surface of complex molecular systems without invoking thermodynamics. J. Chem. Phys., 120:9911–9917, 2004.
S. Curtarolo et al. Crystal structures with data mining of quantum calculations. Phys. Rev. Lett., 91:135503, 2003.
A. R. Oganov, J. Chen, C. Gatti, Y. Ma, Y. Ma, C. W. Glass, Z. Liu, T. Yu, O. O. Kurakevych, and V. L Solozhenko. Ionic high-pressure form of elemental boron. Nature, pages 863–867, 2009.
Y. Ma, M. I. Eremets, A. R. Oganov, Y. Xie, I. Trojan, S. Medvedev, A. O. Lyakhov, M. Valle, and V. Prakapenka. Transparent dense sodium. Nature, 458:182–185, 2009.
A. R. Oganov and S. Ono. Theoretical and experimental evidence for a post-perovskite phase of mgsio3 in earth’s d" layer. Nature, 430:445–448, 2004.
Motohiko Murakami, Kei Hirose, Katsuyuki Kawamura, Nagayoshi Sata, and Yasuo Ohishi. Post-perovskite phase transition in mgsio3. Science, 304:855–858, 2004.
Taku Tsuchiya, Jun Tsuchiya, Koichiro Umemoto, and Renata M. Wentzcovitch. Phase transition in mgsio3 perovskite in the earth’s lower mantle. Earth Planet. Sci. Lett., 224:241 – 248, 2004.
B. Rousseau and N. W. Ashcroft. Observability of a projected new state of matter: a metallic superfluid. Phys. Rev. Lett., 101:046407, 2008.
W. Grochala, R. Hoffmann, J. Feng, and N. W. Ashcroft. The chemical imagination at work in very tight places. Angew. Chem. Int. Ed., 46:3620–3642, 2007.
J. Nagamatsu, N. Nakagawa, T. Muranaka, Y. Zenitani, and J. Akimitsu. Discovery of superconductivity of mgb2 with tc=39k. Nature, 410:63–64, 2001.
P. Prachi. Materials genome initiative and energy. MRS Bulletin, 36:964–966, 2011.
R. M. Martin. Electronic Structure. Cambridge University Press, Cambridge, 2004.
M. S. Woodley, D. P. Battle, D. J. Gale, and R. A. C. Catlow. The prediction of inorganic crystal structures using a genetic algorithm and energy minimisation. Phys. Chem. Chem. Phys., 1:2535–2542, 1999.
A. R. Oganov, A. O. Lyakhov, and M. Valle. How evolutionary crystal structure prediction works - and why. Acc. Chem. Res., 44:227–237, 2011.
K. Yang, W. Setyawan, S. Wang, M. B. Nardeli, and S. Curtarolo. A search model for topological insulators with high-throughput robustness descriptors. Nat. Mater., 5:623–626, 2006.
Q. Li, Y. Ma, A. R. Oganov, H. Wang, H. Wang, Y. Xu, T. Cui, H-K. Mao, and G. Zou. Superhard monoclinic polymorph of carbon. Phys. Rev. Lett., 102:175506, 2009.
C. W. Glass, A. R. Oganov, and N. Hansen. Uspex evolutionary crystal structure prediction. Comput. Phys. Comm., 175:713–720, 2006.
A. R. Oganov and C. W. Glass. Evolutionary crystal structure prediction as a tool in materials design. J. Phys.: Cond. Matter, 20:064210, 2008.
A. R. Oganov and M. Valle. How to quantify energy landscapes of solids. J. Chem. Phys., 130:104504, 2009.
G. H. Jóhannesson, T. Bligaard, A. V. Ruban, H. L. Skriver, K. W. Jacobsen, and J. K. Nørskov. Combined electronic structure and evolutionary search approach to materials design. Phys. Rev. Lett., 88:255506, 2002.
A. O. Lyakhov, A. R. Oganov, H. T. Stokes, and Q Zhu. New developments in evolutionary structure prediction algorithm uspex. Comput. Phys. Comm, 184:1172–1182, 2013.
Q. Zhu, L. Li, A. R. Oganov, and P. B. Allen. Evolutionary prediction of variable stoichiometric surface reconstructions. in preparation, 2013.
Q. Zhu, A. R. Oganov, M. A. Salvadó, P. Pertierra, and A. O. Lyakhov. Denser than diamond:ab initio search for superdense carbon allotropes. Phys. Rev. B, 83:193410, 2011.
A. O. Lyakhov and A. R. Oganov. Evolutionary search for superhard materials: Methodology and applications to forms of carbon and tio . Phys. Rev. B, 84:092103, 2011.
. Phys. Rev. B, 84:092103, 2011.
F. Jensen. Computational Chemistry. Wiley, New York, 1999.
D. A. Case et al. AMBER 12. University of California, San Francisco, 2012.
B. R. Brooks, R. E. Bruccoleri, B. D. Olafson, D. J. States, S. Swaminathan, and M. Karplus. Charmm: A program for macromolecular energy, minimization, and dynamics calculations. J. Comput. Chem., 4:187–217, 1983.
K. Meier, N. Schmid, and V. W. F. Gunsteren. Interfacing the gromos (bio)molecular simulation software to quantum-chemical program packages. J. Comput. Chem., 33:2108–2117, 2012.
M. S. Daw, S. M. Foiles, and M. I. Baskes. The embedded-atom method: a review of theory and applications. Mater. Sci. Rep., 9:251 – 310, 1993.
P. Hohenberg and W. Kohn. Inhomogeneous electron gas. Phys. Rev., 136:B864–B871, 1964.
W. Kohn and L. J. Sham. Self-consistent equations including exchange and correlation effects. Phys. Rev., 140:A1133–A1138, 1965.
J. P. Perdew and A. Zunger. Self-interaction correction to density-functional approximations for many-electron systems. Phys. Rev. B, 23:5048–5079, 1981.
J. P. Perdew et al. Atoms, molecules, solids, and surfaces: Applications of the generalized gradient approximation for exchange and correlation. Phys. Rev. B, 45:13244–13249, 1992.
J. P. Perdew, K. Burke, and M. Ernzerhof. Generalized gradient approximation made simple. Phys. Rev. Lett., 77:3865–3868, 1996.
C. Lee, W. Yang, and R. G. Parr. Development of the colle-salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B, 37:785–789, 1988.
A. D. Becke. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A, 38:3098–3100, 1988.
K. Burke. Perspective on density functional theory. J. Chem. Phys., 136:150901, 2012.
J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou, and K. Burke. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett., 100:136406, 2008.
A. D. Becke. Density-functional thermochemistry. iii. the role of exact exchange. J. Chem. Phys., 98:5648–5652, 1993.
John P. Perdew, Matthias Ernzerhof, and Kieron Burke. Rationale for mixing exact exchange with density functional approximations. J. Chem. Phys., 105:9982–9985, 1996.
A. V. Krukau, O. A. Vydrov, A. F. Izmaylov, and G. E. Scuseria. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys., 125:224106, 2006.
S. Grimme. Semiempirical gga-type density functional constructed with a long-range dispersion correction. J. Comp. Chem., 27:1787–1799, 2006.
M. Dion, H. Rydberg, E. Schröder, D. C. Langreth, and B. I. Lundqvist. Van der waals density functional for general geometries. Phys. Rev. Lett., 92:246401, 2004.
G. Román-Pérez and J. M. Soler. Efficient implementation of a van der waals density functional: Application to double-wall carbon nanotubes. Phys. Rev. Lett., 103:096102, 2009.
J. Klimes, D. R. Bowler, and A. Michaelides. Van der waals density functionals applied to solids. Phys. Rev. B, 83:195131, 2011.
Vladimir I. Anisimov, Jan Zaanen, and Ole K. Andersen. Band theory and mott insulators: Hubbard U instead of stoner I. Phys. Rev. B, 44:943–954, 1991.
D. Vanderbilt. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B, 41:7892–7895, 1990.
P. E. Blochl. Projector augmented-wave method. Phys. Rev. B, 50:17953–17979, 1994.
H. J. Monkhorst and J. D. Pack. Special points for brillouin-zone integrations. Phys. Rev. B, 13:5188–5192, 1976.
J. Behler. Neural network potential-energy surfaces in chemistry: a tool for large-scale simulations. Phys. Chem. Chem. Phys., 13:17930–17955, 2011.
M. C. Payne, M. P. Teter, D. C. Allan, T. A. Arias, and J. D. Joannopoulos. Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients. Rev. Mod. Phys., 64:1045–1097, 1992.
J. D. Gale. General Utility Lattice Program, 2003.
J. D. Gale and A. L. Rohl. The general utility lattice program (gulp). Mol. Simul., 29:291–341, 2003.
S. L. Price et al. Modelling organic crystal structures using distributed multipole and polarizability-based model intermolecular potentials. Phys. Chem. Chem. Phys., 12:8478–8490, 2010.
G. Kresse and J. Furthmüller. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B, 54:11169–11186, 1996.
J. M. Soler et al. The siesta method for ab initio order-n materials simulation. J. Phys.: Cond. Matter, 84:2745, 2002.
K. Schwarz and P. Blaha. Solid state calculations using wien2k. Comput. Mater. Sci., 28:259 – 273, 2003.
A. Togo, F. Oba, and I. Tanaka. First-principles calculations of the ferroelastic transition between rutile-type and 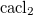 -type
-type 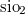 at high pressures. Phys. Rev. B, 78:134106, 2008.
at high pressures. Phys. Rev. B, 78:134106, 2008.
S. Baroni, S. Gironcoli, A. Corso, and P. Giannozzi. Phonons and related crystal properties from density-functional perturbation theory. Rev. Mod. Phys., 73:515–562, 2001.
S. L. Price. The computational prediction of pharmaceutical crystal structures and polymorphism. Adv. Drug Del. Rev., 56:301–319, 2004.
I. A. Baburin, S. Leoni, and G. Seifert. Enumeration of not-yet-synthesized zeolitic zinc imidazolate mof networks: A topological and dft approach. J. Phys. Chem. B, 112:9437–9443, 2008.
J. P. M. Lommerse et al. A test of crystal structure prediction of small organic molecules. Acta Cryst. B, 56:697–714, 2000.
W. D. S. Motherwell et al. Crystal structure prediction of small organic molecules: a second blind test. Acta Cryst. B, B58:647–661, 2002.
G. M. Day et al. A third blind test of crystal structure prediction. Acta Cryst., 61B:511–527, 2005.
G. M. Day et al. Significant progress in predicting the crystal structures of small organic molecules – a report on the fourth blind test. Acta Cryst., B65:107–125, 2009.
D. A. Bardwell et al. Towards crystal structure prediction of complex organic compounds – a report on the fifth blind test. Acta Cryst., B67:535–551, 2011.
S. Kim et al. Crystal structure prediction of flexible molecules using parallel genetic algorithms with a standard force field. J. Comput. Chem, 30:1973–1985, 2009.
P. Raiteri, R. Martonak, and M. Parrinello. Exploring polymorphism: the case of benzene. Angew. Chem. Int. Ed., 44:3769–3773, 2005.
G. M. Day. Current approaches to predicting molecular organic crystal structures. Cryst. Rev., 17:3–52, 2011.
A. L. Lyakhov, A. R. Oganov, and M. Valle. How to predict very large and complex crystal structures. J. Comput. Phys. Comm., 181:1623–1632, 2010.
C. P. Brock and J. D. Dunitz. Towards a grammar of crystal packing. Chem. Mater., 6:1118–1127, 1994.
W. H. Baur and D. Kassner. The perils of cc: comparing the frequencies of falsely assigned space groups with their general population. Acta Cryst., B48:356–369, 1992.
R. Hoft, J. D. Gale, and M. J. Ford. Implementation of a z-matrix approach within the siesta periodic boundary conditions code and its application to surface adsorption. Mol. Simul., 32:595–600, 2006.
Y. Li et al. Van der waals interactions in molecular assemblies from first-principles calculations. J. Phys. Chem. A, 114:1944–1952, 2010.
H. Fukazawa, S. Ikeda, and S. Mae. Incoherent inelastic neutron scattering measurements on ice xi; the proton-ordered phase of ice ih doped with koh. Chem. Phys. Lett., 282:215–218, 1998.
A. J. Leadbetter et al. The equilibrium low-structure of ice. J. Chem. Phys., 82:424–428, 1985.
B. J. Murray, D. A. Knopf, and A. K. Bertram. The formation of cubic ice under conditions relevant to earth’s atmosphere. Nature, 434:202–205, 2005.
K. Umemoto, R. M. Wentzcovitch, S. Saito, and T. Miyake. Body-centered tetragonal c4: A viable carbon allotrope. Phys. Rev. Lett., 104:125504, 2010.
X. F. Zhou et al. Ab initio study of the formation of transparent carbon under pressure. Phys. Rev. B, 82:134126, 2010.
J. Yang et al. ice tessellation on a hydroxylated silica surface. Phys. Rev. Lett., 92:146102, 2005.
W. B. Hubbard et al. Interior structure of neptune: comparison with uranus. Science, 253:648–651, 1991.
R. Bini and G. Pratesi. High-pressure infrared study of solid methane: Phase diagram up to 30 gpa. Phys. Rev. B, 55:14800, 1997.
H. E. Maynard-Casely et al. The distorted close-packed crystal structure of methane a. J. Chem. Phys., 133:064504, 2010.
I. Nakahata et al. Structural studies of solid methane at high pressures. Chem. Phys. Lett., 302:359–362, 1999.
L. Sun et al. X-ray diffraction studies and equation of state of methane at 202gpa. Chem. Phys. Lett., 473:72–74, 2009.
G. Gao et al. Dissociation of methane under high pressure. J. Chem. Phys., 133:144508, 2010.
A. D. Fortes et al. Ab initio simulation of ammonia monohydrate (nh3-h2o) and ammonium hydroxide (nh4oh). J. Chem. Phys., 115:7006, 2001.
F. Datchi et al. Solid ammonia at high pressure: A single-crystal x-ray diffraction study to 123gpa. Phys. Rev. B, 73:17411, 2006.
J. S. Loveday et al. Structure of deuterated ammonia iv. Phys. Rev. Lett, 76:74–77, 1996.
C. J. Pickard and R. J. Needs. Highly compressed ammonia forms an ionic crystal. Nature Mater., 7:775–779, 2008.
M. Santoro and F. A. Gorelli. High pressure solid state chemistry of carbon dioxide. Chem. Soc. Rev., 35:918–931, 2008.
B. Olinger. The compression of solid co at 296 k to 10 gpa. J. Chem. Phys., 77:6255–6258, 1982.
N. Tajima et al. First principles prediction of crystal structures of co2. Electron. J. Theor. Chem., 2:139–148, 1997.
B. Holm et al. Theoretical investigation of high pressure phases of carbon dioxide. Phy. Rev. Lett., 85:1258, 2000.
A. R. Oganov et al. Novel high-pressure structures of mgco3, caco3 and co2 and their role in the earth’s lower mantle. Earth Planet. Sci. Lett., 273:38–47, 2008.
C. S. Yoo et al. Crystal structure of carbon dioxide at high pressure: superhard polymeric carbon dioxide. Phy. Rev. Lett., 83:5527–5530, 1999.
S. A. Bonev et al. High-pressure molecular phases of solid carbon dioxide. Phy. Rev. Lett., 91:065501, 2003.
M. M. Thiery and J. M. Leger. High pressure solid phases of benzene. i. raman and x-ray studies of c6h6 at 294 k up to 25 gpa. J. Chem. Phys., 89:4255–4271, 1988.
L. Ciabini et al. High-pressure and high-temperature equation of state and phase diagram of solid benzene. Phys. Rev. B, 72:094108, 2005.
L. Ciabini et al. Triggering dynamics of the high-pressure benzene amorphization. Nat. Mater., 6:39–43, 2007.
David Van Der Spoel, Erik Lindahl, Berk Hess, Gerrit Groenhof, Alan E. Mark, and Herman J. C. Berendsen. Gromacs: Fast, flexible, and free. J. Comput. Chem., 26(16):1701–1718, 2005.
X. D. Wen, R. Hoffmann, and N. W. Ashcroft. Benzene under high pressure: a story of molecular crystals transforming to saturated networks, with a possible intermediate metallic phase. J. Am. Chem. Soc., 133:9023–9035, 2011.
J. A. Chisholm et al. An ab initio study of observed and hypothetical polymorphs of glycine. Cryst. Growth. Des., 5:1437–1442, 2005.
S. Hamad, C. E. Hughes, and C. Richard. Clustering of glycine molecules in aqueous solution studied by molecular dynamics simulation. J. Phys. Chem. B, 112:7280–7288, 2008.
A. Dawson et al. Effect of high pressure on the crystal structures of polymorphs of glycine. Cryst. Growth. Des., 5:1415–1427, 2005.
S. A. Moggach, S. Parsons, and P. A. Wood. High-pressure polymorphism in amino acids. Cryst. Rev., 14:143–184, 2008.
V. Boldyreva, T. N. Drebushchak, and E. S. Shutova. Structural distortion of the  ,
, 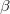 and
and 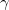 polymorphs of glycine on cooling. Z. Kristallogr, 218:366–376, 2003.
polymorphs of glycine on cooling. Z. Kristallogr, 218:366–376, 2003.
G. He et al. Direct growth of -glycine from neutral aqueous solutions by slow, evaporation-driven crystallization. Crys. Growth Des., 6:1746–1749, 2006.
G. L. Pervolich, L. K. Hansen, and A. Bauer-Brandl. The polymorphism of glycine. thermochemical and structural aspects. J. Thermal Anal. Calorimetry, 66:699–715, 2001.
C. Blerk and G. J. Kruger. Butane-1,4-diammonium dibromide. Acta. Cryst., E63:o342–o344, 2007.
M. Ji, C. Wang, and K. M. Ho. Comparing efficiencies of genetic and minima hopping algorithms for crystal structure prediction. Phys. Chem. Chem. Phys., 12:11617–11623, 2010.
J. L. Hoard, R. E. Hughes, and D. E. Sands. The structure of tetragonal boron. J. Am. Chem. Soc., 80:4507–4515, 1958.
M. A. Neumann and M. Perrin. The computational prediction of pharmaceutical crystal structures and polymorphism. J. Phys. Chem. B, 109:15531, 2005.
L. Schlapbach and A. Zuttel. Hydrogen-storage materials for mobile applications. Nature, 414:353–358, 2001.
B. C. Wood and N. Marzari. Dynamics and thermodynamics of a novel phase of naalh4. Phys. Rev. Lett., 103:185901, 2009.
A. Tekin, R. Caputo, and A. Züttel. First-principles determination of the ground-state structure of libh4. Phys. Rev. Lett., 104:215501, 2010.
Y. Filinchuk, B. Richter, T. R. Jensen, V. Dmitriev, D. Chernyshov, and H. Hagemann. Porous and dense magnesium borohydride frameworks: Synthesis, stability, and reversible absorption of guest species. Angew. Chem. Int. Ed., 50:11162–11166, 2011.
Q. Zhu, A. R. Oganov, C. W. Glass, and H. T. Stokes. Constrained evolutionary algorithm for structure prediction of molecular crystals: methodology and applications. Acta Cryst. B, 68:215–226, 2012.
L. George, V. Drozd, S. K. Saxena, E. G. Bardaji, and M. Fichtner. Structural phase transitions of mg(bh4)2 under pressure. J. Phys. Chem. C, 113:15087–15090, 2009.
R. Cerny, Y. Filinchuk, H. Hagemann, and K. Yvon. Magnesium borohydride: Synthesis and crystal structure. Angew. Chem. Int. Ed., 46:5765–5767, 2007.
J. Her et al. Structure of unsolvated magnesium borohydride mg(bh4)2. Acta Cryst., B63:561–568, 2007.
A. Bil, B. Kolb, R. Atkinson, D. G. Pettifor, T. Thonhauser, and A. N. Kolmogorov. van der waals interactions in the ground state of mg(bh4)2 from density functional theory. Phys. Rev. B, 83:224103, 2011.
R. Cerny, D. B. Ravnsbak, P. Schouwink, Y. Filinchuk, N. Penin, J. Teyssier, L. Smrcok, and T. R. Jensen. Potassium zinc borohydrides containing triangular [zn(bh4)3]- and tetrahedral [zn(bh4)xcl4-x]2- anions. J. Phys. Chem. C, 116:1563–1571, 2012.
V. Ozolins, E. H. Majzoub, and C. Wolverton. First-principles prediction of a ground state crystal structure of magnesium borohydride. Phys. Rev. Lett., 100:135501, 2008.
J Voss, J S Hummelshøj, Z Łodziana, and T Vegge. Structural stability and decomposition of mg(bh4)2 isomorphsan ab initio free energy study. J. Phys. Cond. Matt., 21:012203, 2009.
X. F. Zhou et al. Crystal structure and stability of magnesium borohydride from first principles. Phys. Rev. B, 79:212102, 2009.
J. Fan, K. Bao, D. F. Duan, L. C. Wang, B. B. Liu, and T. Cui. High volumetric hydrogen density phases of magnesium borohydride at high-pressure: A first-principles study. Chin. Phys. B, 21:086104, 2012.
G. Henkelman, Arnaldsson A., and H. Jonsson. A fast and robust algorithm for bader decomposition of charge density. Comput. Mater. Sci., 36:354 – 360, 2006.
F. Birch. Elasticity and constitution of the earth’s interior. J. Geophys. Res., 57:227, 1952.
H. A. Levy and P. A. Agron. The crystal and molecular structure of xenon difluoride by neutron diffraction. J. Am. Chem. Soc., 85:241–242, 1963.
D. H. Templeton, A. Zalkin, J. D. Forrester, and S. M. Williamson. Crystal and molecular structure of xenon trioxide. J. Am. Chem. Soc., 85:817–817, 1963.
S. Hoyer, T. Emmler, and K. Seppelt. The structure of xenon hexafluoride in the solid state. J. Fluorine Chem., 127:1415–1422, 2006.
M. Kim, M. Debessai, and C-S Yoo. Two- and three-dimensional extended solids and metallization of compressed xef2. Nat. Chem., 2:784–788, 2010.
M. Somayazulu, P. Dera, A. F. Goncharov, S. A. Gramsch, P. Liermann, W. Yang, Z. Liu, H-K Mao, and R. J. Hemley. Pressure-induced bonding and compound formation in xenon-hydrogen solids. Nat. Chem., 2:50–53, 2010.
D. F. Smith. Xenon trioxide. J. Am. Chem. Soc., 85:816–817, 1963.
H. Selig, H. H. Claassen, C. L. Chernick, J. G. Malm, and J. L. Huston. Xenon tetroxide: Preparation and some properties. Science, 143:1322–1323, 1964.
D. S. Brock and G. J. Schrobilgen. Synthesis of the missing oxide of xenon, xeo2, and its implications for earthâmissing xenon. J. Am. Chem. Soc., 133:6265–6269, 2011.
W. Grochala. Atypical compounds of gases, which have been called ’noble’. Chem. Soc. Rev., 36:1632–1655, 2007.
E. Anders and T. Owen. Mars and earth: Origin and abundance of volatiles. Science, 198(4316):453–465, 1977.
C Sanloup, R. J. Hemley, and H-K Mao. Evidence for xenon silicates at high pressure and temperature. Geophys. Res. Lett., 29:1883–1886, 2002.
C. Sanloup, B. C. Schmidt, E. M. C. Perez, A. Jambon, E. Gregoryanz, and M. Mezouar. Retention of xenon in quartz and earth’s missing xenon. Science, 310:1174–1177, 2005.
A. R. Oganov, Y Ma, C. W. Glass, and M Valle. Evolutionary crystal structure prediction: overview of the uspex method and some of its applications. Psi-K Newsletter, 84:142–171, 2007.
W. A. Caldwell, J. H. Nguyen, B. G. Pfrommer, F. Mauri, S. G. Louie, and R. Jeanloz. Structure, bonding, and geochemistry of xenon at high pressures. Science, 277:930–933, 1997.
R. Dovesi, V. R. Saunders, C. Roetti, R. Orlando, C. M. Zicovich-Wilson, F. Pascale, B. Civalleri, K. Doll, N. M. Harrison, I. J. Bush, Ph. D’Arco, and M. Llunell. CRYSTAL06 User’s Manual. University of Torino, 2006.
M. D. Towler, N. L. Allan, N. M. Harrison, V. R. Saunders, W. C. Mackrodt, and E. Aprà. Ab initio study of mno and nio. Phys. Rev. B, 50:5041–5054, 1994.
K. A. Peterson, D Figgen, E Goll, H Stoll, and M Dolg. Systematically convergent basis sets with relativistic pseudopotentials. ii. small-core pseudopotentials and correlation consistent basis sets for the post-d group 16?18 elements. J. Chem. Phys., 119:11113–11123, 2003.
C. Gatti. TOPOND-98: An Electron Density Topological Program for Systems Periodic in N (N=0-3) Dimensions, User’s Manual. CNR-ISTM, Milano, 1999.
C. Gatti. chapter 7, The Quantum Theory of Atoms in Molecules: From Solid State to DNA and Drug Design. Wiley-VCH, 2007.
T. Keith. Molecules in Magnetic Fields. PhD thesis, McMaster University, 1993.
M. Shishkin and G. Kresse. Self-consistent 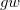 calculations for semiconductors and insulators. Phys. Rev. B, 75:235102, 2007.
calculations for semiconductors and insulators. Phys. Rev. B, 75:235102, 2007.
V. S. Urusov. Theory of Isomorphous Miscibility. Nauka, Moscow, 1977.
Y. Ma, A. R. Oganov, and C. W. Glass. Structure of the metallic 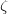 -phase of oxygen and isosymmetric nature of the
-phase of oxygen and isosymmetric nature of the  - phase transition: Ab initio simulations. Phys. Rev. B, 76:064101, 2007.
- phase transition: Ab initio simulations. Phys. Rev. B, 76:064101, 2007.
L. F. Lundegaard, G. Weck, M. I. McMahon, S. Desgreniers, and P. Loubeyre. Observation of an o8 molecular lattice in the [epsiv] phase of solid oxygen. Nature, 443:201–204, 2006.
D. R. Sears and H. P. Klug. Density and expansivity of solid xenon. J. Chem. Phys., 37:3002–3006, 1962.
Y. Sonnenblick, E. Alexander, Z.H. Kalman, and I.T. Steinberger. Hexagonal close packed krypton and xenon. Chem. Phys. Lett., 52:276 – 278, 1977.
A. D. Becke and K. E. Edgecombe. A simple measure of electron localization in atomic and molecular systems. J. Chem. Phys., 92:5397–5403, 1990.
R. F. W. Bader. Atoms in Molecules - A Quantum Theory. Oxford University Press, 1990.
D. J. Frost, C. Liebske, F. Langenhorst, C. A. McCammon, R. G. Tronnes, and D. C. Rubie. Experimental evidence for the existence of iron-rich metal in the earth’s lower mantle. Nature, 428:409–412, 2004.
F. W. Zhang and Oganov A. R. Valence state and spin transitions of iron in earth’s mantle silicates. Earth. Planet. Sci. Let., 249:436 – 443, 2006.
J. Lin, D. L. Heinz, H-K. Mao, R. J. Hemley, J. M. Devine, J. Li, and G. Shen. Stability of magnesiowüstite in earth’s lower mantle. Proc. Nat. Aca. Sci., 100:4405–4408, 2003.
I. I. Mazin, Y. Fei, R. Downs, and R. E. Cohen. Possible polytypism in feo at high pressures. American Mineralogist, 83:451–457, 1998.
A. R. Oganov, R. Martonak, A. Laio, P. Raiteri, and Parrinello M. Anisotropy of earth’s d” layer and stacking faults in the mgsio3 post-perovskite phase. Nature, 438:1142–1144, 2005.
V. S. Urusov and V. B. Dudnikova. The trace-component trapping effect: Experimental evidence, theoretical interpretation, and geochemical applications. Geochimica et Cosmochimica Acta, 62:1233 – 1240, 1998.
Thomas S. Duffy, Russell J. Hemley, and Ho-kwang Mao. Equation of state and shear strength at multimegabar pressures: Magnesium oxide to 227 gpa. Phys. Rev. Lett., 74:1371–1374, 1995.
M. J. Mehl, Cohen R. E., and H. Krakauer. Linearized augmented plane wave electronic structure calculations for mgo and cao. J. GeoPhys. Res., 118:8009 – 8022, 1988.
A. R. Oganov, M. J. Gillan, and G. D. Price. Ab initio lattice dynamics and structural stability of mgo. J. Chem. Phys., 118:10174–10182, 2003.
A. B. Belonoshko, S. Arapan, R. Martonak, and A. Rosengren. Mgo phase diagram from first principles in a wide pressure-temperature range. Phys. Rev. B, 81:054110, 2010.
Koichiro Umemoto, Renata M. Wentzcovitch, and Philip B. Allen. Dissociation of mgsio3 in the cores of gas giants and terrestrial exoplanets. Science, 311:983–986, 2006.
H. Wriedt. The mgo (magnesium-oxygen) system. J. Phase Equilibria, 8:227–233, 1987.
J. M. Recio and Ravindra Pandey. Ab initio study of neutral and ionized microclusters of mgo. Phys. Rev. A, 47:2075–2082, 1993.
Z.L. Wang, J. Bentley, E.A. Kenik, L.L. Horton, and R.A. McKee. In-situ formation of mgo2 thin films on mgo single-crystal surfaces at high temperatures. Surf. Sci., 273:88 – 108, 1992.
Q. Zhu, D. Y. Jung, A. R. Oganov, C. W. Glass, C. Gatti, and A. O. Lyakhov. Stability of xenon oxides at high pressures. Nature Chem., 5:61–65, 2013.
E. Zurek, R. Hoffmann, N. W. Ashcroft, A. R. Oganov, and A. O. Lyakhov. A little bit of lithium does a lot for hydrogen. Proc. Natl. Aca. Sci., 106:17640–17643, 2009.
Ross T. Howie, Olga Narygina, Christophe L. Guillaume, Shaun Evans, and Eugene Gregoryanz. High-pressure synthesis of lithium hydride. Phys. Rev. B, 86:064108, 2012.
W. Zhang, A. R. Oganov, A. F. Goncharov, Q. Zhu, S. E. Boulfelfel, A. O. Lyakhov, M. Somayazulu, and V. B. Prakapenka. Unexpected stable stoichiometries of sodium chlorides. arXiv preprint arXiv:1211.3644, 2012.
Andriy O. Lyakhov, Artem R. Oganov, Harold T. Stokes, and Qiang Zhu. New developments in evolutionary structure prediction algorithm uspex. Comp. Phys. Comm., 184:1172 – 1182, 2013.
N. Vannerberg. Progress in Inorganic Chemistry. John Wiley Sons, Inc., 2007.
S. C. Abrahams and J. Kalnajs. The formation and structure of magnesium peroxide. Acta Cryst., 7:838–842, 1954.
I. Efthimiopoulos, K. Kunc, S. Karmakar, K. Syassen, M. Hanfland, and G. Vajenine. Structural transformation and vibrational properties of bao2 at high pressures. Phys. Rev. B, 82:134125, 2010.
N. G. Vannerberg. The formation and structure of magnesium peroxide. Ark. Kemi, 14:99–105, 1959.
H. Olijnyk and W. B. Holzapfel. High-pressure structural phase transition in mg. Phys. Rev. B, 31:8412–4683, 1985.
R. M. Wentzcovitch and M. L. Cohen. Theoretical model for the hcp-bcc transition in mg. Phys. Rev. B, 37:5571–5576, 1988.
P. Li, G. Gao, Y. Wang, and Y. Ma. Crystal structures and exotic behavior of magnesium under pressure. J. Phys. Chemi. C, 114:21745–21749, 2010.
R. D. Shannon and C. T. Prewitt. Effective ionic radii in oxides and fluorides. Acta Cryst., B25:925–946, 1969.
J. T. Waber and Cromer D. T. Orbital radii of atoms and ions. J. Chem. Phys., 42:4116–4123, 1965.
Aliaksandr V. Krukau, Oleg A. Vydrov, Artur F. Izmaylov, and Gustavo E. Scuseria. Influence of the exchange screening parameter on the performance of screened hybrid functionals. J. Chem. Phys., 125:224106, 2006.
K. Tanigaki et al. Superconductivity at 33 k in cs rb
rb c60. Nature, 352:222–223, 1991.
c60. Nature, 352:222–223, 1991.
E. A. Ekimov et al. Superconductivity in diamond. Nature, 428:542–545, 2004.
J. C. Mayer, A. K. Geim, M. I. Katsnelson, K. S. Novoselov, T. J. Booth, and S. Roth. The structure of suspended graphene sheets. Nature, 446:60, 2007.
W-L. Mao, H-K. Mao, P. J. Eng, T. P. Trainor, M. Newville, C. Kao, D. L. Heinz, J. Shu, Y. Meng, and R. J. Hemley. Bonding changes in compressed superhard graphite. Science, 302:425–427, 2003.
Wang. Z. W. et al. A quenchable superhard carbon phase synthesized by cold compression of carbon nanotubes. Proc. Natl. Acad. Sci., 101:13699–13702, 2004.
C. J. Pickard and R. J. Needs. Hypothetical low-energy chiral framework structure of group 14 elements. Phys. Rev. B, 81:014106, 2010.
R. Hoffmann, T. Hughbanks, and M. Kertesz. Hypothetical metallic allotrope of carbon. J. Am. Chem. Soc., 105:4831–4832, 1983.
V. V. Brazhkin. Interparticle interaction in condensed media: some elements are ‘more equal than others’. Phys. Usp., 52:369, 2009.
M. D. Knudson, M. P. Desjarlais, and D. H. Dolan. Shock-wave exploration of the high-pressure phases of carbon. Science, 302:1822–1825, 2008.
R. L. Johnston and R. Hoffmann. Superdense carbon, c8: supercubane or analog of .gamma.-silicon? J. Am. Chem. Soc., 111:810–819, 1989.
R. Biswas, Richard M. Martin, R. J. Needs, and O. H. Nielsen. Stability and electronic properties of complex structures of silicon and carbon under pressure: Density-functional calculations. Phys. Rev. B, 35:9559–9568, 1987.
S. J. Clark, G. J. Ackland, and J. Crain. Theoretical stability limit of diamond at ultrahigh pressure. Phys. Rev. B, 52:15035–15038, 1995.
A. Wosylus, Y. Prots, W. Schnelle, M. Hanfland, and U. Schwarz. Crystal structure refinements of ge (tp12), physical properties and pressure-induced phase transformation ge (tp12) to ge (ti4). J. Chem. Sci, 63:608 – 614, 2008.
C. T. Prewitt and H. S. Young. Germanium and silicon disulfides: Structure and synthesis. Science, 149:535–537, 1965.
B. J. Skinner and D. E. Appleman. Melanophlogite, a cubic polymorph of silica. Am. Mineral, 48:854, 1963.
D. Bakowies, A. Gelessus, and W. Thiel. Quantum chemical study of c78 fullerene isomers. Chem. Phys. Lett., 11:324–329, 1992.
Th. Frauenheim, G. Jungnickel, Th. Kohler, and U. Stephan. Structure and electronic properties of amorphous carbon: from semimetallic to insulating behaviour. J. Non-Cryst. Solids, 182:186 – 197, 1995.
F. Gao et al. Hardness of covalent crystals. Phys. Rev. Lett., 91:015502, 2003.
F. Occelli, P. Loubeyre, and R. Letoullec. Properties of diamond under hydrostatic pressures up to 140 gpa. Nat. Mater., 2:151–154, 2003.
M. Shishkin, M. Marsman, and G. Kresse. Accurate quasiparticle spectra from self-consistent gw calculations with vertex corrections. Phys. Rev. Lett., 99:246403, 2007.
J. Paier, M. Marsman, and G. Kresse. Dielectric properties and excitons for extended systems from hybrid functionals. Phys. Rev. B, 78:121201, 2008.
M. Cardona and M. L. W. Thewalt. Isotope effects on the optical spectra of semiconductors. Rev. Mod. Phys., 77:1173–1224, 2005.
L. D. Landau, E. M. Lifshitz, and L. P. Pitaevskii. Electrodynamics of Continuous Media. Butterworth-Heinermann, Oxford, UK, 1984.
A. Laio and M. Parrinello. Escaping free-energy minima. Proc. Natl. Acad. Sci., 99:12562–12566, 2002.
R. Martonak, D. Donadio, A. R. Oganov, and M. Parrinello. Crystal structure transformations in sio2 from classical and ab initio metadynamics. Nat. Mater., 5:623–626, 2006.
M. Parrinello and A. Rahman. Crystal structure and pair potentials: A molecular-dynamics study. Phys. Rev. Lett., 45:1196–1199, 1980.
Arthur F. Voter. A method for accelerating the molecular dynamics simulation of infrequent events. J. Chem. Phys., 106:4665–4677, 1997.
K. Li, X. Wang, F. Zhang, and D. Xue. Electronegativity identification of novel superhard materials. Phys. Rev. Lett., 100:235504, 2008.
R. Martonak, A. Laio, M. Bernasconi, C. Ceriani, P. Raiteri, F. Zipoli, and M. Parrinello. Simulation of structural phase transitions by metadynamics. Z. Kristallogr., 220:489–498, 2005.
S. F. Pugh. Relations between elastic moduli and plastic properties of polycrystalline pure metals. Philos. Mag., 45:823–843, 1954.
B. W. H. Beest, G. J. Kramer, and R. A. Santen. Force fields for silicas and aluminophosphates based on ab initio calculations. Phys. Rev. Lett., 64:1955–1958, 1990.
A. R. Oganov, J. P. Brodholt, and G. D Price. Comparative study of quasiharmonic lattice dynamics, molecular dynamics and debye model in application to mgsio3 perovskite. Phys. Earth Planet. Inter., 122:277 – 288, 2000.
A. R. Oganov, G. D. Price, and J. P. Brodholt. Theoretical investigation of metastable al2sio5 polymorphs. Acta Cryst. A, 57:548–557, 2001.
V. S. Urusov, V. R. Oganov, and N. N. Eremin. Computer simulation of the structure, properties, and stability of the al2sio5 polymorphs: I. ionic model. Geochem. Int., 36:3897–414, 1998.
S. E. Boulfelfel, A. R. Oganov, and S. Leoni. Understanding the nature of “superhard graphite". Sci. Rep., 2:471, 2012.
T. Irifune et al. Materials: Ultrahard polycrystalline diamond from graphite. Nature, 421:599–600, 2003.
R. B. Aust and H. G. Drickamer. Carbon: A new crystalline phase. Science, 140:817–819, 1963.
M. Hanfland, K. Syassen, and R. Sonnenschein. Optical reflectivity of graphite under pressure. Phys. Rev. B, 40:1951–1954, 1989.
Y. Zhao and I. L. Spain. X-ray diffraction data for graphite to 20 gpa. Phys. Rev. B, 40:993–997, 1989.
W. Utsumi and T. Yagi. Light-transparent phase formed by room-temperature compression of graphite. Science, 252:1542–1544, 1991.
J. Wang, C. Chen, and Y. Kawazoe. Low-temperature phase transformation from graphite to 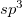 orthorhombic carbon. Phys. Rev. Lett., 106:075501, 2011.
orthorhombic carbon. Phys. Rev. Lett., 106:075501, 2011.
D. Selli, I. A. Baburin, R. Martonak, and S. Leoni. Superhard sp3 carbon allotropes with odd and even ring topologies. Phys. Rev. B, 84:161411, 2011.
Z. Zhao et al. Novel superhard carbon: C-centered orthorhombic 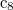 . Phys. Rev. Lett., 107:215502, 2011.
. Phys. Rev. Lett., 107:215502, 2011.
M. Amsler et al. The crystal structure of cold compressed graphite. Phys. Rev. Lett., 108:065501, 2012.
H. Niu et al. Families of superhard crystalline carbon allotropes constructed via cold compression of graphite and nanotubes. Phys. Rev. Lett., 108:135501, 2012.
R. H. Baughman, A. Y. Liu, C. Cui, and P. J. Schields. A carbon phase that graphitizes at room temperature. Synthetic Metals, 86:2371 – 2374, 1997.
V. Greshnyakov and E. Belenkov. Structures of diamond-like phases. JETP, 113:86–95, 2011.
Q. Zhu, A. R. Oganov, and A. O. Lyakhov. Evolutionary metadynamics: a novel method to predict crystal structures. CrystEngComm, 14:3596–3601, 2012.
| Crystal Structure Prediction and its Application in Earth and Materials Sciences |