| Crystal Structure Prediction and its Application in Earth and Materials Sciences |
| Crystal Structure Prediction and its Application in Earth and Materials Sciences |
Evolutionary crystal structure prediction pioneered in the works of Oganov et al proved to be a powerful approach in determining the crystal structure of materials. This thesis mainly covers two subjects of crystal structure prediction: 1) Methodology development; 2) Application to earth and materials sciences.
First of all, we describe how to predict crystal structure by evolutionary approach, and extend this method to study the packing of organic molecules, by our specially designed constrained evolutionary algorithm. The main feature of this new approach is that each unit or molecule is treated as a whole body, which drastically reduces the search space and improves the efficiency. The improved method is possibly to be applied in the fields of (1) high pressure phase of simple molecules (H O, NH
O, NH , CH
, CH , etc); (2) pharmaceutical molecules (glycine, aspirin, etc); (3) complex inorganic crystals containing cluster or molecular unit, (Mg(BH), Ca(BH), etc).
, etc); (2) pharmaceutical molecules (glycine, aspirin, etc); (3) complex inorganic crystals containing cluster or molecular unit, (Mg(BH), Ca(BH), etc).
One application of the constrained evolutionary algorithm is given by the study of (Mg(BH), which is a promising materials for hydrogen storage. Our prediction does not only reproduce the previous work on Mg(BH) at ambient condition, but also yields two new tetragonal structures at high pressure, with space groups 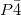 and
and 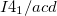 are predicted to be lower in enthalpy, by 15.4 kJ/mol and 21.2 kJ/mol, respectively, than the earlier proposed
are predicted to be lower in enthalpy, by 15.4 kJ/mol and 21.2 kJ/mol, respectively, than the earlier proposed 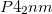 phase. We have simulated X-ray diffraction spectra, lattice dynamics, and equations of state of these phases. The density, volume contraction, bulk modulus, and the simulated XRD patterns of and structures are in excellent agreement with the experimental results.
phase. We have simulated X-ray diffraction spectra, lattice dynamics, and equations of state of these phases. The density, volume contraction, bulk modulus, and the simulated XRD patterns of and structures are in excellent agreement with the experimental results.
Materials under pressure often exhibit exotic physical and chemical behaviors. In particular, extremely new stable compounds appear. Here, we focus on the variation of stoichiometry under pressure. Two kinds of oxides (Xe-O and Mg-O) have been studied under megabar pressures. For XeO, we predict the existence of thermodynamically stable Xe-O compounds at high pressures (XeO, XeO and XeO become stable at pressures of 83, 102 and 114 GPa, respectively). For Mg-O, our calculations find that two extraordinary compounds MgO and MgO become thermodynamically stable at 116 GPa and 500 GPa, respectively. Our calculations indicate large charge transfer in these oxides for both systems, suggesting that large electronegativity difference and pressure are the key factors favoring their formations. We also discuss if these oxides might exist at earth and planetary conditions.
If the target properties are set as the global fitness functions while structure relaxations are energy/enthalpy minimization, such hybrid optimization technique could effectively explore the landscape of properties for the given systems. Here we illustrate this function by the case of searching for superdense carbon allotropes. We find three structures (hP3, tI12, and tP12) that have significantly greater density. Furthermore, we find a collection of other superdense structures based on different ways of packing carbon tetrahedral. Superdense carbon allotropes are predicted to have remarkably high refractive indices and strong dispersion of light.
Apart from evolutionary approach, there also exist some other methods for structural prediction. One can also combine the features from different methods. We develop a novel method for crystal structure prediction, based on metadynamics and evolutionary algorithms. This technique can be used to produce efficiently both the ground state and metastable states easily reachable from a reasonable initial structure. We use the cell shape as collective variable and evolutionary variation operators developed in the context of the USPEX method to equilibrate the system as a function of the collective variables. We illustrate how this approach helps one to find stable and metastable states for AlSiO , SiO, MgSiO. Apart from predicting crystal structures, the new method can also provide insight into mechanisms of phase transitions. This method is especially powerful in sampling the metastable structures from a given configuration. Experiments on cold compression indicated the existence of a new superhard carbon allotrope. Numerous metastable candidate structures featuring different topologies have been proposed for this allotrope. We use evolutionary metadynamics to systematically search for possible candidates which could be accessible from graphite. Our calculation not only found all the previous proposed candidates for superhard graphite, but also predicted two allotropes (X-carbon and Y-carbon) showing unusual types of 5+7 and 4+8 topologies. These superhard carbon allotropes can be classified into five families based on 6 (diamond/lonsdaleite), 5+7 (M- and W-carbon), 5+7 (X-carbon), 4+8 (bct-C4), and 4+8 (Y-carbon) topologies.
, SiO, MgSiO. Apart from predicting crystal structures, the new method can also provide insight into mechanisms of phase transitions. This method is especially powerful in sampling the metastable structures from a given configuration. Experiments on cold compression indicated the existence of a new superhard carbon allotrope. Numerous metastable candidate structures featuring different topologies have been proposed for this allotrope. We use evolutionary metadynamics to systematically search for possible candidates which could be accessible from graphite. Our calculation not only found all the previous proposed candidates for superhard graphite, but also predicted two allotropes (X-carbon and Y-carbon) showing unusual types of 5+7 and 4+8 topologies. These superhard carbon allotropes can be classified into five families based on 6 (diamond/lonsdaleite), 5+7 (M- and W-carbon), 5+7 (X-carbon), 4+8 (bct-C4), and 4+8 (Y-carbon) topologies.
| Crystal Structure Prediction and its Application in Earth and Materials Sciences |